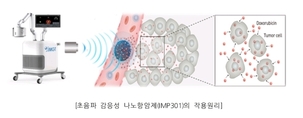
아이엠지티(대표 이학종)는 나노입자 기술을 이용, 집속초음파에 반응하여 항암제 성분이 방출되도록 설계된 초음파 감응성 나노항암제의 국내 임상시험계획을 식약처로부터 지난 3일 승인받았다고 밝혔다. 이번 1상 임상 시험에서는 표준요법에 실패한 진행성, 또는 전이성 고형암 환자를 대상으로 항암제인 독소루비신을 봉입한 초음파 감응성 나노입자(IMP301)의 안전성, 내약성을 평가하고, 2상 임상시험에서의 권장용량을 결정하게 된다. 현재 아이엠지티는 항암제 침투력 개선 목적의 집속초음파시스템(IMD10)을 개발하여 췌장암 환자를 대상으로 국내 임상시험을 진행 중인데, 초음파 감응성 나노항암제는 암조직에 전달되어 축적되고, 아이엠지티사의 집속초음파 시스템에 반응하여 표적 암조직내에서 항암제 성분이 방출되고 침투되도록 설계된 혁신적 약물전달 모델이다. 독소루비신은 심기능 이상 등 전신 부작용에도 불구하고, 연조직골육종, 난소암, 유방암등 치료옵션이 제한적인 여러 난치성 고형암에 치료약제로 사용되고 있다. 아이엠지티가 개발한 초음파 감응성 나노약물 전달 모델을 이용하면, 리포좀 봉입을 통해 독소루비신의 전신 부작용을 줄일 수 있을 뿐 아니라, 표적 종양에서의 전달력 개선을

샤페론(378800)이 아토피 치료제(이하 누겔)의 미국 임상2상 시험계획서를 미국 식품의약국(이하 FDA)에 제출했다고 31일 밝혔다. 샤페론은 이번 다국가 임상 2상에서 경증 또는 중등증의 아토피 피부염 환자 210명을 대상으로 약동학, 안전성, 내약성, 유효성을 평가할 계획이다. 대상환자는 이중 눈가림 방식으로 위약과 누겔을 약 8주간 바르게 된다. 샤페론은 이번 임상을 통해 아토피 피부염 환자에 대한 위약 대비 누겔의 습진 범위 및 중증도 지수(EASI 점수) 개선 효과 확인을 목표로 하고 있다. 누겔은 면역 및 혈관 세포에 존재하는 염증복합체를 억제해 아토피 피부염 증상을 악화하는 사이토카인 발현을 낮추는 신약 후보 물질로서, 체내 염증 조절 세포 수를 증가시켜 이중으로 광범위한 염증 병리 기전을 제어한다.

큐라클(365270, 대표 유재현)은 궤양성 대장염(Ulcerative Colitis) 치료제 신약 후보물질 ‘CU104’의 임상 2상 IND(임상시험계획)를 동유럽 3개국 각 의약품 규제당국에 제출했다고 17일 공시를 통해 밝혔다. 큐라클은 지난 6월 미국 FDA로부터 CU104 임상 2상 IND를 승인받았으며, 이번에는 유럽 임상을 진행하기 위해 동유럽 보스니아, 세르비아, 마케도니아 각국의 의약품 규제당국과 충분한 사전 논의를 거쳐 IND를 제출했다. 큐라클은 CU104 임상을 미국, 유럽, 한국 등에서 글로벌 임상으로 준비하고 있다. 향후 한국 식품의약품안전처 IND 승인을 거쳐 다국가 임상에 본격 진입할 방침이라고 회사 측은 전했다.

지엔티파마(대표이사 곽병주)는 퇴행성 뇌신경질환 치료제로 개발 중인 ‘크리스데살라진’의 임상 2상 시험계획서(IND)를 식품의약품안전처에 제출했다고 12일 밝혔다. 이번 임상 2상은 인지기능장애를 겪고 있으면서 뇌 아밀로이드 양전자 단층촬영(PET) 영상에서 양성으로 확인된 중등도 알츠하이머병 환자 144명을 대상으로 진행한다. 대상 환자는 이중 눈가림 방식으로 위약과 크리스데살라진 100mg, 200mg을 1일 1회, 26주 동안 복용하게 된다. 1차 유효성 평가는 26주째 알츠하이머병 평가 척도(ADAS-cog) 점수가 개선된 환자의 비율로 크리스데살라진의 약효를 검증한다. 2차 유효성 평가는 13주, 26주째에 인지기능, 일상생활능력, 신경정신행동, 노인우울척도 점수의 변화로 위약 대비 크리스데살라진의 약효를 확인한다. 임상시험 책임자는 인하대병원 신경과 최성혜 교수이며 국내외 10여개 치매 임상기관이 참여한다.크리스데살라진은 과학기술정보통신부 뇌프론티어 사업단의 지원을 받아 치매 치료제로 발굴한 합성신약으로, 활성산소를 제거하는 강력한 항산화작용과 mPGES-1을 억제해 염증인자인 PGE2 생성을 차단하는 소염작용을 동시에 갖고 있다. 비임상시험에서
티움바이오(KOSDAQ: 321550)는 중국 파트너사 한소제약(Hansoh Pharmaceutical)이 중국 국가약품관리감독국(NMPA)으로부터 자궁내막증 신약 후보물질 ‘TU2670’(한소제약 코드명 HS-10518)의 임상1상에 대한 임상시험계획서(IND)를 승인받았다고 5일 밝혔다. ‘TU2670’은 자궁내막증 및 자궁근종 질환 치료영역에서 가장 주목받고 있는 경구용 GnRH(성선자극호르몬 분비호르몬) 길항제(antagonist) 기전의 신약으로, 한소제약은 작년 8월 티움바이오와 1억7천만 달러(약 2210억 원) 규모의 라이선스 계약을 통해 중국지역에 대한 ‘TU2670’의 개발 및 상업화 권리를 보유하고 있다. 이번 승인된 임상은 한소제약이 중국에서 진행하는 TU2670의 임상1상시험이고, 건강한 가임기 여성 48명을 대상으로 용량 증량 시험을 통해 TU2670의 내약성 및 안전성 등을 평가할 예정이다.
대웅제약(대표 전승호·이창재)은 보툴리눔 톡신 ‘나보타(미국명 주보)’가 미국에서 미간주름 환자를 대상으로 실시한 6개월 장기지속 효과를 입증하기 위한 고용량 임상 2상을 성공적으로 완료했다고 4일 밝혔다. 이번 임상은 대웅제약의 미국 파트너사 에볼루스(Evolus)가 65세 미만의 미간주름 중등도 내지 중증 이상의 150명의 환자를 대상으로 작년 3월부터 다기관, 이중맹검 및 무작위 방식으로 12개월동안 진행됐다. 이번 연구에서 고용량 40유닛 투여 시 6개월 또는 26주의 장기지속 효과를 확인했으며, 심각한 부작용 없이 안정성이 입증됐다고 밝혔다. 40유닛 주보의 활성 대조군은 20유닛 보톡스 및 20유닛 주보로, 활성 대조군과의 부작용 결과값은 유사했다.

코넥스트는 미국 FDA로부터 듀피트렌구축 치료제인 CNT201의 임상 1/2상 임상시험계획(이하 IND)을 승인 받았으며 올해 하반기부터 본격적인 글로벌 임상개발에 착수한다고 26일 밝혔다. 지난 5월 FDA에 IND를 신청한 지 1개월 만에 승인을 획득하였으며, 총 60명의 듀피트렌구축(Dupuytren’s contracture) 환자들을 대상으로 CNT201의 안전성과 유효성 및 약동학 등에 관한 연구를 진행할 계획이다.

유한양행(대표이사 조욱제)은 만성 자발성/유발성 두드러기, 아토피 피부염, 알레르기 천식, 식품 알레르기 등 면역글로불린 E (IgE)가 매개된 다양한 알레르기 질환 치료용 신약으로 개발 중인 YH35324의 임상 1a상 파트A 결과를 유럽 알레르기임상면역학회(EAACI)에서 6월 10일(토) 발표하였다. YH35324는 Fc 융합단백질 신약으로, 혈중 유리 IgE 수준을 낮추어 알레르기 증상을 개선시키는 작용을 한다. 유럽 알레르기임상면역학회는 매년 전 세계 알레르기질환 전문가 1만여명이 참석하는 권위 있는 학술대회다. 국내 4개 대학병원 알레르기내과에서 진행한 임상시험결과를 EAACI 2023년 연례 회의의 포스터 발표 세션에서 아주대학교병원 알레르기내과 예영민 교수가 발표하였다. 이 임상시험은 YH35324를 사람에게 처음으로 투여하는(First-In-Human, FIH) 임상 1a상 파트A 시험으로, 아토피가 있는 건강인 또는 경증의 알레르기 질환 환자에게 YH35324를 단계적인 용량 증량 방식으로 단회 투여한 후 안전성, 약동학, 약력학적 특성을 평가하였다. 아주대학교병원 예영민 교수는, “YH35324 모든 용량에서 우수한 내약성과 안전성이 관찰
바이엘은 비트락비(성분명: 라로트렉티닙.사진)가 4건의 연구 분석 결과를 통해 NTRK 유전자 융합 암 성인 및 소아환자 대상 장기 효능 및 안전성 프로파일을 입증했다고 밝혔다. 해당 결과는 지난 2일부터 6일까지 진행된 2023년 미국임상종양학회(ASCO) 연례 학술대회에서 공개되었다. -TRK 융합 암 성인 환자 대상 라로트렉티닙의 장기 효능 및 안전성(Abstract 3141) NTRK 유전자 융합이 발견된 비중추신경계 성인 암환자 194명 중 적합 환자 180명을 대상으로 한 장기간의 추적연구(데이터 컷오프 2022년 7월)의 하위그룹 분석 최신 결과에서 라로트렉티닙의 안전성 프로파일이 입증되었다. IRC(Independent Review Committee)에 따른 평가 가능 환자의 객관적 반응률은 57%(95% CI 50-65)로, 완전관해 16%(병리학적 완전관해 1건 포함), 부분관해 41%를 기록했다. 중추신경계 전이가 있는 평가 가능 환자(n=22)의 경우, 객관적 반응률은 68%(95% CI 45-86)로 나타났다. 추적기간 중앙값 32.3개월 시점에서 전체 환자의 치료반응 도달 기간(time to response)의 중앙값은 1.8개월,
mRNA 치료제 및 백신 분야 선도 바이오테크 기업 모더나는 19일(현지시각) 자사의 프로피온산혈증(Propionic acidemia, PA) mRNA 치료제 후보물질 mRNA-3927의 임상 1/2상 중간 결과를 2023 미국 유전자세포치료학회(American Society of Gene + Cell Therapy(ASGCT) Annual Meeting))에서 발표했다. 진행 중인 글로벌 임상 1/2상은 프로피온산혈증이 유전자적으로 확인된 1세 이상의 참가자를 대상으로 mRNA-3927의 안전성과 약리 및 약동성을 평가하기 위한 오픈 라벨, 다기관 용량 최적화 연구이다. (ClinicalTrials.gov Identifier: NCT04159103) 이 연구는 mRNA-3927의 정맥 내 투여를 평가하기 위해 용량 증량 접근법을 사용한다. 초기 투여 요법으로 0.3mg/kg을 3주마다 정맥 내 투여했으며 후속 용량은 2주마다 투여됐다. 용량 최적화 시험(10회 용량)을 완료한 참가자는 오픈 라벨 연구(NCT05130437)에서 치료를 계속할 수 있는 자격이 부여된다. 1차 평가 변수는 안전성과 내약성이며, 2차 및 탐색적 평가 변수는 약리, 잠재적 혈장 바
(주) 메디팜헬스뉴스/등록번호 서울 아 015222/등록일자 2011년 2울 23일/제호 메디팜헬스/발행인 김용발/편집인 노재영/발행소 서울특별시 송파구 송파대로 42길 45 메디팜헬스빌딩 1층
발행일자 2011년 3월 3일/청소년보호 책임자 김용발/Tel. 02-701-0019 / Fax. 02-701-0350 /기사접수 imph7777@naver.com
메디팜헬스뉴스의 모든 기사는 저작권의 보호를 받습니다. 따라서 무단사용하는 경우 법에 저촉됩니다.
UPDATE: 2024년 05월 04일 06시 30분








