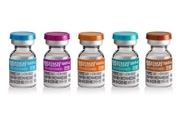
JW중외제약은 A형 혈우병 치료제 ‘헴리브라(성분명 에미시주맙)’를 투약한 환자의 다양한 신체활동 데이터와 안전성을 입증한 연구 결과가 최근 국제학술지 ‘International Journal of Hematology (Int J Hematol)’에 게재됐다고 18일 밝혔다. 헴리브라는 혈우병 환자의 몸속에 부족한 혈액응고 제8인자를 모방하는 혁신 신약이다. A형 혈우병 치료제 중 유일하게 기존 치료제(8인자 제제)에 대한 내성을 가진 항체 환자와 비항체 환자 모두 사용할 수 있다. 최대 4주 1회 피하주사로 예방 효과가 지속되는 특징도 있다. 지난해 5월에는 건강보험 급여 대상이 만 1세 이상의 비항체 중증 A형 혈우병 환자로 확대됐다. 일본 나라의과대학 소아과 케이지 노가미(Keiji Nogami) 교수 연구팀은 지난 2019년 1월부터 2021년 5월까지 평균 연령 35세인 비항체 A형 혈우병 환자 107명을 대상으로 연구를 진행했다. 연구팀은 전자 환자보고 애플리케이션 ‘ePRO’와 착용형 활동추적기(Wearable activity tracker)를 통해 헴리브라 투약 후 환자들의 신체활동과 출혈 여부, 안전성 등의 연관관계를 평가했다. 연구결과 6세

카나리아바이오는 16일오후11시에 DSMB(Data Safety Monitoring Board)가 신규난소암 환자를 대상으로진행하고 있는 오레고보맙글로벌 임상3상의무용성 평가를 진행하였고임상 지속을 위한 P value를 달성하지못해 임상시험 중단을권고했다고 밝혔다. 다만 DSMB는면역항암제의 특성상 전체생존기간에서 유의미한 효능을보일 가능성이 있다는의견을 제시하고 추적관찰은지속할 것을 권고했다. 카나리아바이오 나한익 대표는 “임상 2상의결과와 상반된 결과에대한 원인분석을 통해추후 계획을 세우도록하겠다”고말했다. 카나리아바이오는 100% 자회사카나리아바이오(구 MHC&C)를 통해임상개발을 진행하고 있다.

LG화학이 HPV(human papillomavirus, 인유두종 바이러스) 음성 두경부암 환자들의 치료 기회 확대를 위한 임상 3상에 본격 착수한다. LG화학은 17일, 미국 항암신약 개발사 ‘아베오(AVEO Pharmaceuticals)’가 두경부암 신약물질인 ‘파이클라투주맙(Ficlatuzumab)’의 미국 임상 3상(시험명 FIERCE-HN)을 본격화하며, 첫 시험자를 등록했다고 밝혔다. ‘아베오’는 LG화학의 손자회사 편입 후 신장암 치료제 ‘포티브다(FOTIVDA)’를 이을 후속 항암제 개발에 속도를 내고 있다. ‘파이클라투주맙’은 종양을 키우는 간세포 성장인자(HGF, hepatocyte growth factor)의 작용을 억제하는 기전의 단일항체 기반 표적항암제이다. LG화학은 이번 임상 3상에서 두경부암 치료에 쓰이는 표적항암제 ‘얼비툭스(ERBITUX®, 성분명 cetuximab)’ 단일 요법을 대조군으로 ‘파이클라투주맙’ 및 ‘얼비툭스’ 병용 요법의 유효성 및 안전성을 평가할 계획이다. 이전에 백금 기반 항암화학요법과 면역관문억제제를 단일 요법으로 순차적 투약했거나 병용 투약했던 HPV 음성(negative) 두경부암 환자 중 암의 악화,

JW중외제약은 말레이시아 국립의약품규제기관(NPRA)으로부터 통풍치료제 에파미뉴라드(코드명 URC102)에 대한 임상 3상 시험계획(IND)을 승인받았다고 15일 밝혔다. 에파미뉴라드의 임상 3상은 한국을 포함한 아시아 5개국에서 총 588명의 통풍 환자를 대상으로 페북소스타트 대비 유효성(혈중 요산 감소 효과)과 안전성을 평가하는 시험이다. 이번 승인에 따라, JW중외제약은 에파미뉴라드의 다국가 임상 3상 IND를 모두 승인받았다. 한국에서는 2022년 11월 IND 승인 후 지난해 3월 환자 등록 및 투약을 시작했으며, 대만에서도 2023년 8월 IND 승인 후 12월 첫 환자 등록을 마쳤다. 이어 9월 태국과 싱가포르에서 IND를 승인받았다. 경구제로 개발하고 있는 에파미뉴라드는 hURAT1(human uric acid transporter-1)을 선택적으로 저해하는 기전의 요산 배설 촉진제로, 혈액 내에 요산 농도가 비정상적으로 높은 고요산혈증 및 통풍질환에 유효한 신약후보물질이다. JW중외제약은 지난 2021년 3월 종료된 국내 임상 2b상에서 에파미뉴라드의 우수한 내약성과 안전성을 확인했으며 1차와 2차 유효성 평가변수도 모두 충족했다.
압타머 플랫폼 전문 기업 압타머사이언스(코스닥 291650, 대표이사 한동일)는 첫 번째 신약 파이프라인인 AST-201에 대한 식품의약품안전처 IND 승인을 위해 필요한 준비를 모두 마쳤다고 11일 밝혔다. AST-201은 간암이나 폐암 환자의 암세포에 많이 발현되는 GPC3 수용체를 표적으로 한 ApDC (Aptamer -Drug Conjugate)계 약물로, GPC3에 선택적으로 결합하는 압타머에 항암제로 사용되는 젬시타빈을 접합시킨 물질이다. 젬시타빈은 췌장암, 폐암 치료 목적으로 광범위하게 사용되는 약물이지만 약물 분해효소가 많이 존재하는 간암의 경우 치료 효과를 보이지 않았으나, 압타머와 결합시킨 ApDC 형태의 AST-201에서는 간암 세포에 대한 선택적 표적 전달 및 체내 안정성을 기반으로 우수한 약효를 보여줘 효과적인 약물이 많지 않은 간암 치료에 새로운 옵션을 제시할 전망이다.

-삼성바이오에피스 위반 내역 - 삼성바이오에피스가 야심차게 준비하고 있는 황반변성 치료제 개발에 복병이. 생겼다.식품의약품안전처는 최근 삼성바이오에피스의 SB15(성분명 애플리버셉트)와 아일리아간 임상시험에 대해. 약사법 위반 혐의를 적용 업무정지 1개월 15일 처분을 내렸다. 이에따라 삼성바이오임상시는 내년 2월2일까지 해당 제품의 임상을. 진행할 수. 없게돼. 이들 품목을. 개발하고. 임상에 나선 셀트리온 등 경쟁사 보다 불리한 시간에 놓이게 됐다. 해당 임상시험은 SB15와 아일리아 간 유효성, 안전성, 약동학 및 면역원성을 비교하는 제3상, 무작위 배정, 이중 눈가림, 평행군, 다기관 임상시험 등이다.
SK바이오사이언스와 사노피는 공동 개발중인 21가 폐렴구균 단백접합 백신후보물질 ‘GBP410(사노피 과제명 ‘SP0202’)의 미국(FDA) 임상 3상 IND(시험계획)를 현지 시간 8일 제출하며 임상에 돌입하기 위한 절차에 착수했다고 11일 밝혔다. FDA 임상 3상 IND 신청 절차는 앞으로 추가적인 제출 과정을 거쳐 완료될 예정이다. 앞서 지난 8월 SK바이오사이언스와 사노피는 GBP410의 임상2상을 성공적으로 마친 바 있다. 생후 12~15개월 소아 140명과 42~89일 영유아 712명을 대상으로 진행한 GBP410과 대조백신(프리베나 13)의 비교 임상에서 GBP410은 대조백신 대비 동등한 수준의 면역원성을 확인했다. 안전성 측면에서 GBP410 접종군은 백신과 관련 있는 중대한 이상사례가 보고되지 않았다. 또 파상풍, 디프테리아, 백일해, 폴리오, B형 헤모필루스 인플루엔자 백신 등 영유아 및 소아 접종 권고 백신을 병용 투약하는 경우에도 대조백신대비 동등한 수준의 면역원성 및 안전성을 확인했다. SK바이오사이언스와 사노피는 이 같은 임상2상 결과를 토대로 미국, 유럽, 한국 등 다국가 영유아를 대상으로 2027년 내 임상3상까지 완료한다는
바이오스플라이스 테라퓨틱스(Biosplice Therapeutics)는 무릎 골관절염 치료신약 후보물질 ‘로어시비빈트(Lorecivivint)’에 대한 임상 3상의 장기 연장시험인 ‘OA-07’ 결과를 2023년 11월13일 캘리포니아 샌디에고에서 개최된 미국 류마티스학회(ACR)에서 구두 발표했다. ‘로어시비빈트’는 미국 샌디에고 소재 바이오텍인 ‘바이오스플라이스 테라퓨틱스(Biosplice Therapeutics)’가 개발중인 무릎 골관절염 치료신약 후보물질이다. 지난 2021년 3월 삼일제약(000520)이 국내 허가 및 판매에 대한 독점권리를 확보한 바 있다. ‘로어시비빈트’는 ‘CLK/DYRK 키나제(인산화효소)’ 억제제로 ‘Wnt 신호’를 조절하는 기전을 갖고 있어 세계 최초의 골관절염 근본적 치료제(DMOAD ; Disease-Modifying Osteoarthritis Drug)를 목표로 개발되고 있다. ‘OA-07’은 미국에서 12개월 동안 임상 3상인 ‘OA-11’을 완료한 환자중 276명을 대상으로 진행한 장기 연장시험으로 ‘로어시비빈트’를 매년 1회씩 3년간 투여했을 때 ‘구조 개선 가능성(structure improvement pote

동아에스티(대표이사 사장 김민영)는 지난 24일 식품의약품안전처로부터 면역항암제 ‘DA-4505’의 임상 1/2a상 시험계획(IND)을 승인받았다고 27일 밝혔다. 이번 임상은 국소 진행성 또는 전이성 고형암 성인 환자를 대상으로 DA-4505 단독요법 또는 항 PD-1 면역관문억제제 Pembrolizumab 병용요법의 안전성, 내약성, 약동학, 약력학 및 유효성을 평가한다. DA-4505는 AhR(Aryl Hydrocarbon Receptor, 아릴탄화수소수용체) 길항제다. AhR은 면역계를 조절하는 인자로, 면역반응을 억제하고 종양 세포가 공격받는 것을 방지하는 역할을 한다. 전임상에서 DA-4505는 AhR을 저해함으로써 종양미세환경에서 억제된 면역반응을 복구시켰다. 또한 수지상세포, T세포 등 자극성 면역세포를 활성화시키고 암세포가 면역을 억제하는 기능을 감소시켰다.특히, 글로벌제약사가 개발 중인 AhR 길항제와 비교하는 전임상을 통해 개선된 종양 억제 효과를 확인했다. DA-4505와 항 PD-1 면역관문억제제인 Pembrolizumab와 병용투여를 통해 증대된 종양 억제 효과를 확인했다.
에스바이오메딕스(304360, 공동대표 김동욱, 강세일)는 중증하지허혈 (CLI, Critical Limb Ischemia) 세포치료제 FECS-Ad (동종지방유래 중간엽줄기세포 스페로이드)의 임상 제1상 및 2a상 환자 투여를 완료했다고 13일 밝혔다. 임상시험은 삼성서울병원 혈관외과 김동익 교수 주도 하에 진행 중으로 치료제를 투여 받은 환자는 임상시험계획에 따라 6개월 간 안전성 및 유효성을 관찰하고 평가하게 된다. 마지막 임상시험 대상자에 대한 투여가 완료됨에 따라 6개월 간 추적 관찰 이후 임상시험 결과를 도출하며, 최종 임상시험 종료는 내년 중순경으로 예상된다. 중증하지허혈은 하지혈관의 협착, 폐색 또는 폐쇄로 인한 혈류의 감소로 점진적인 하지허혈이 발생하고 심한 허혈성 통증을 유발, 조직의 괴사 등을 일으키는 질병이다. 동맥경화성 말초동맥질환 (PAD, Peripheral Artery Disease)의 가장 심각한 임상 양상 중 하나이며, 치료가 지연될 시 6개월 내 주요 사지를 절단하는 상황을 초래할 수도 있다. 일반적인 관상동맥 질환과 마찬가지로 40세 이후 발병하기 시작하여 나이가 들수록 발병률이 증가한다. 당뇨병 환자에게서 많이 발생하는
(주) 메디팜헬스뉴스/등록번호 서울 아 015222/등록일자 2011년 2울 23일/제호 메디팜헬스/발행인 겸 편집인 노재영/발행소 서울특별시 송파구 송파대로 42길 45 메디팜헬스빌딩 1층
발행일자 2011년 3월 3일/청소년보호 책임자 노재영/Tel. 02-701-0019 / Fax. 02-701-0350 /기사접수 imph7777@naver.com
메디팜헬스뉴스의 모든 기사는 저작권의 보호를 받습니다. 따라서 무단사용하는 경우 법에 저촉됩니다.
UPDATE: 2026년 04월 22일 09시 30분







